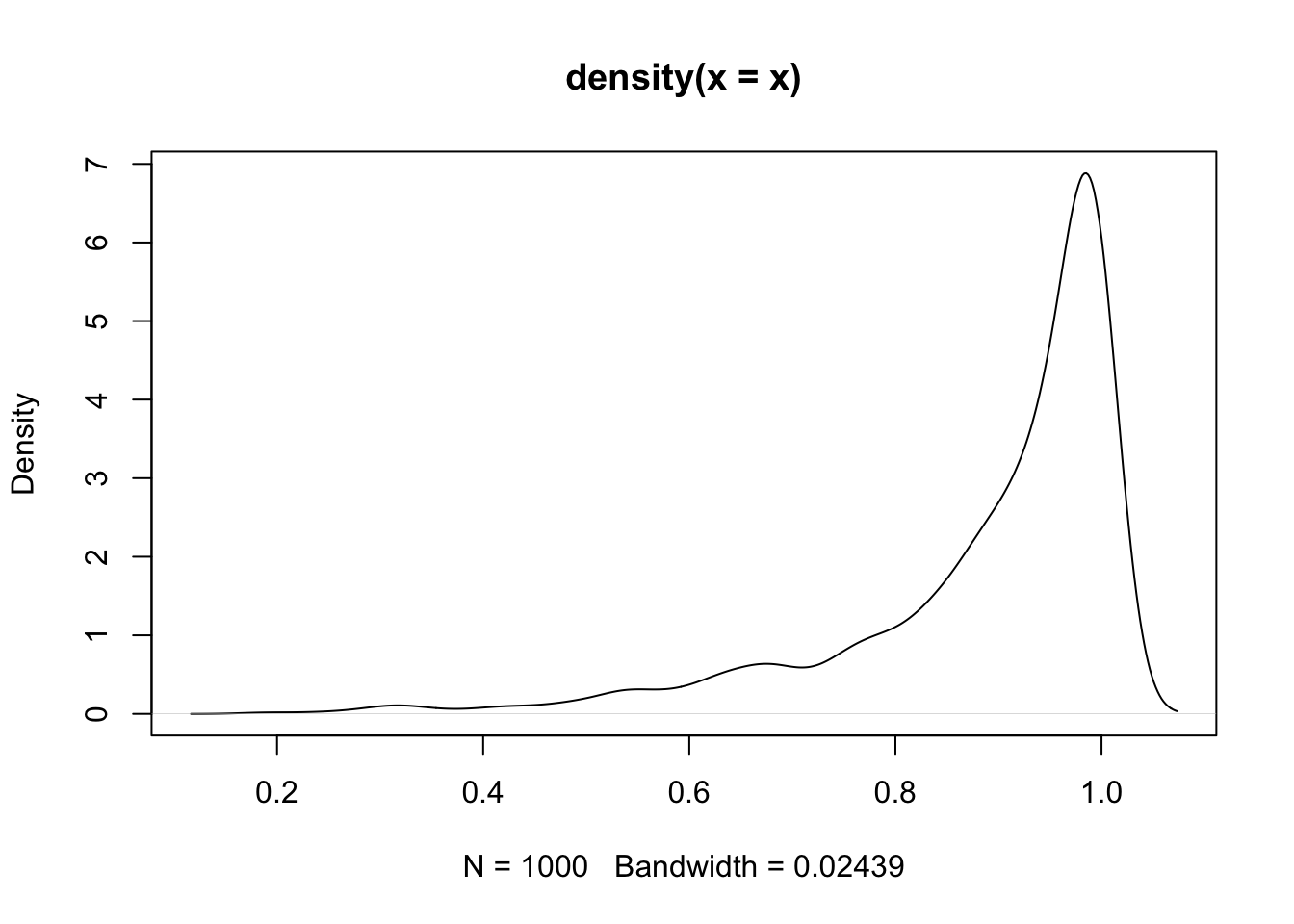
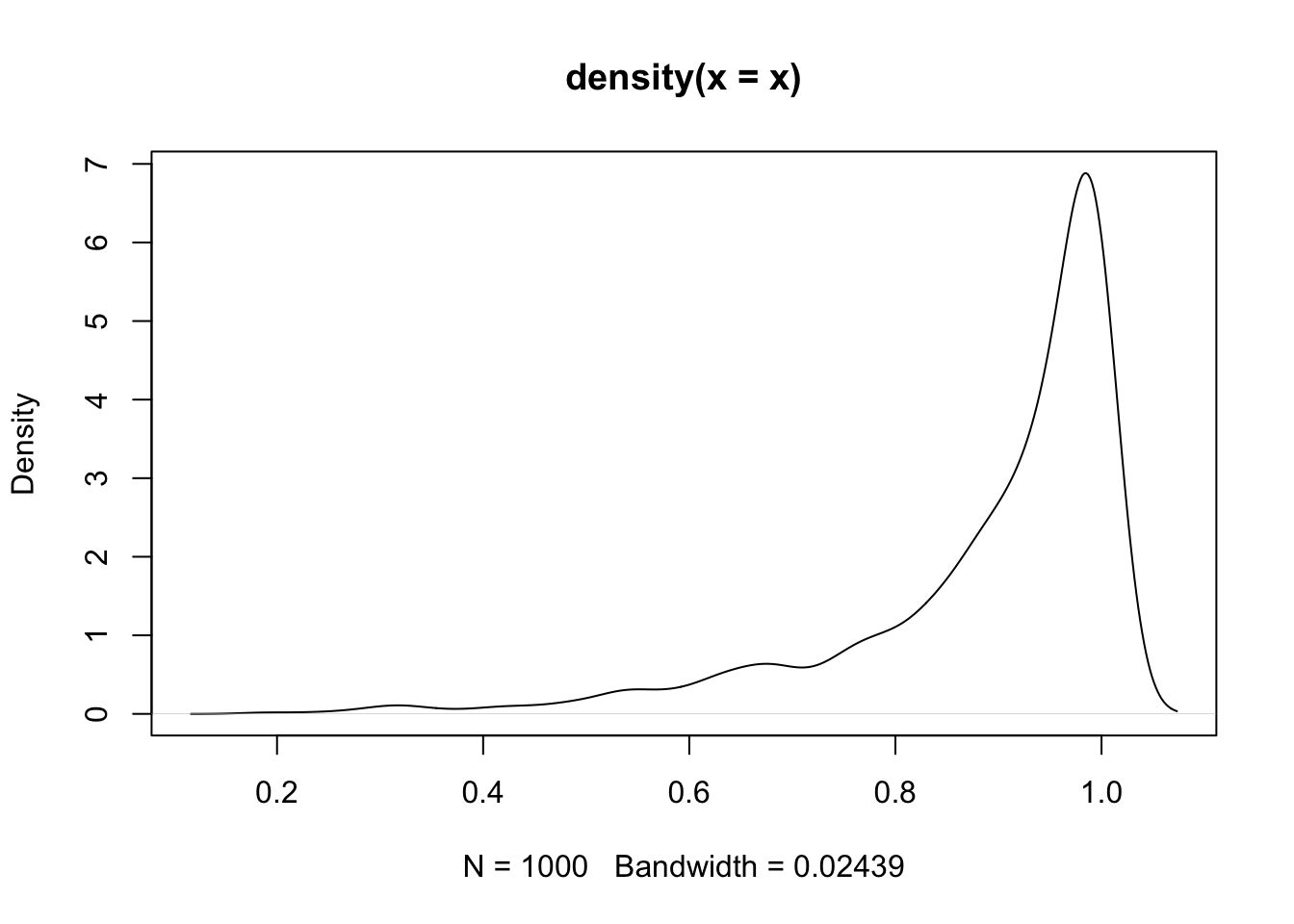
nasal <- read_csv("https://raw.githubusercontent.com/ChristopherCarignan/journal-articles/refs/heads/master/rtMRI-velum/velum_data.csv") |>
filter(
stress == "N",
vowel %in% c("a_", "E_", "I_", "O_", "U_")
) |>
mutate(
voicing = case_when(
post %in% c("nt__", "nt_@", "nt_6", "nt_a") ~ "voiceless",
post %in% c("nd_@", "nd_6", "nd_a") ~ "voiced"
),
vowel = str_to_lower(vowel) |> str_remove("_")
) |>
# drop codas not included in the analysis
drop_na(voicing) |>
mutate(
nas_dur = (Vokal_off - velumopening_maxvel_on) * 1000,
nas_prop = nas_dur / (Vokal_dur * 1000),
NC = ifelse(voicing == "voiceless", "nt", "nd")
) |>
# drop observations with wrong fMRI tracking
filter(nas_prop > 0, nas_prop < 1) |>
select(
speaker, label, vowel, NC, voicing, nas_prop
)